State of Wall in Protein Language Models in 2026
Last year Pascal Notin wrote a great post summarizing important observation about AI + proteins: Have we hit the scaling wall for protein language models?. (Spoiler: the answer is ‘yes’)
Briefest summary if you didn’t read it:
- PLMs’ performance on fitness prediction (‘transferability’ of skills) plateaus after 1B and declines after 5B parameters. This holds for multiple PLM families
- leading approaches combine MSAs and 3d structure. Even very simple methods that combine these sources of information outperform billion-parameter models
- training on genetic sequences (that’s quite a lot of additional signal!) doesn’t help — Evo and Evo-2 are near the bottom of the leaderboard
Remark: I’ll focus on sequence-based models, and declare folding and inverse folding as out-of-scope for this post.
New models appeared on ProteinGym leaderboard since Pascal’s post, but conclusions hold. And later analysis from another group corroborates this: Medium-sized PLMs perform well at transfer learning on realistic datasets. Folding models keep using embeddings from (very old) ESM-2.
We’re in a weird position when we have a lot of sequencing data (and computing power), but we can’t put it to work. Let’s take a tour across recent literature and see if there are any signs of going beyond this scaling wall.
Remark: for comparison, widely used structure models (AlphaFold2 / AlphaFold3 / proteinMPNN) are even less than 1B parameters. This could be explained by a smaller size of PDB compared to UniProt, or maybe it’s just a common trait of molecular biology.
AMPLIFY: is scaling necessary?
Interestingly, the authors explicitly start from noting that the premise that “scale leads to performance” is likely false in PLMs, and then use recent LLM pretraining techniques to achieve better perplexity than ESM-2 using a cheaper and smaller model.
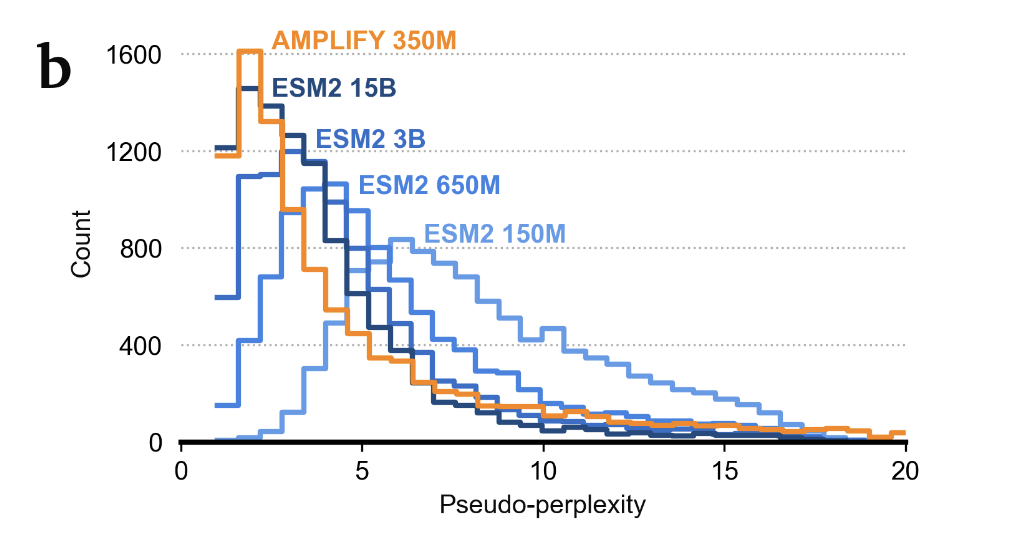
They explore removal of UniProt clustering (used in most models) to increase size/diversity of training data. Their main argument: clustering adds too much weight to non-realistic sequences.
Validation, interestingly, is a subset of human proteome — choice here is important because final ranking in perplexity is highly affected by similarity of distribution to training data.
Turns out, quality of sequencing data matters a lot — significant improvements correlate with largest “clean-ups” in UniProt.
Other interesting bits:
- AF2 can’t distinguish between non-proteins and disordered proteins (PLMs of course can)
- sequence recovery is very good (a lot of analysis in supplements)
- analysis of performance on downstream tasks (like protein properties) is lacking, but this was covered in other papers.
Overall: yes, we can significantly improve perplexity/recovery, and model size isn’t crucial.
Structure-alignment of ESM2 and AMPLIFY
Multiple works in this list sprinkle structure tokens in training (and sometimes inference). This work instead utilizes a CLIP-like contrastive alignment step between PLM token and protein GNN (GearNet) structure token. Second loss is a direct prediction of structure tokens.
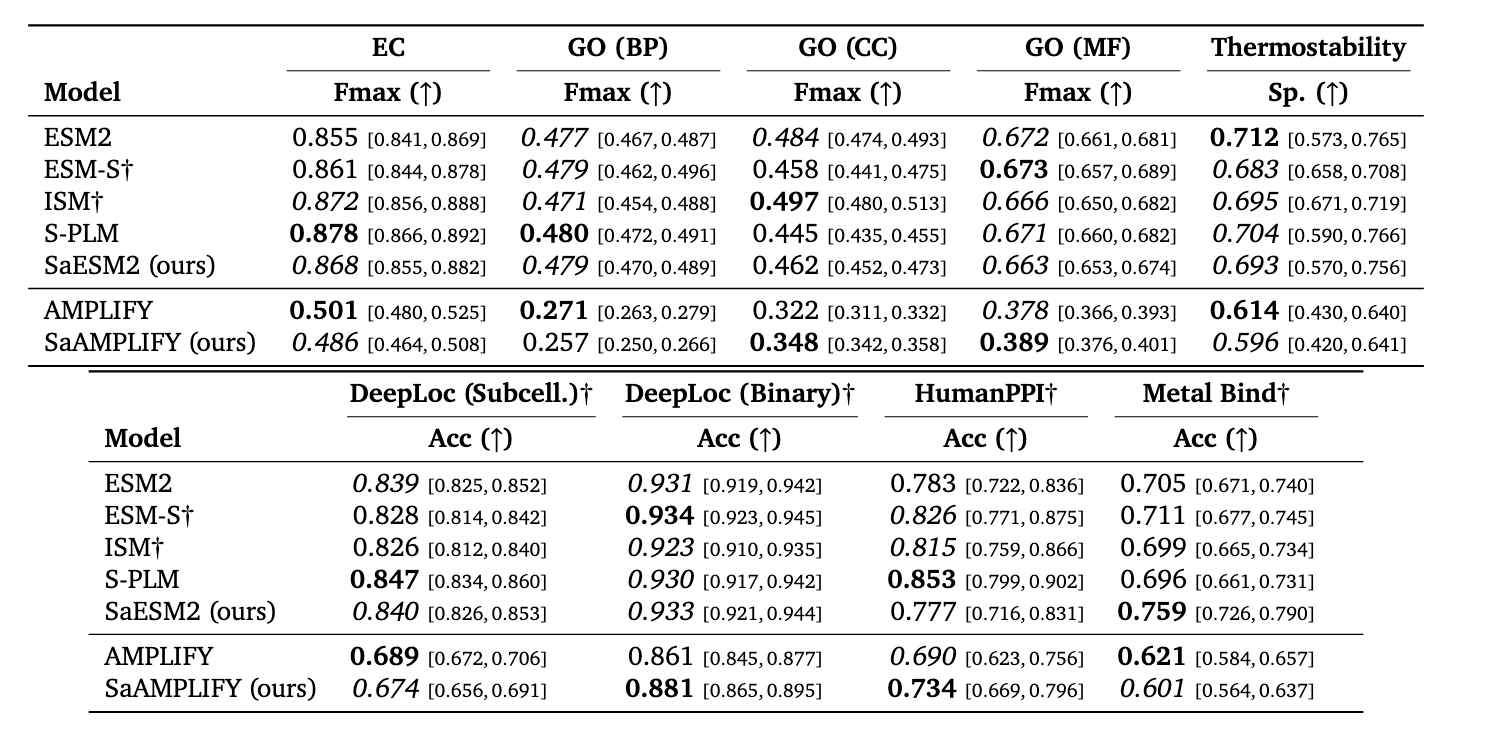
This delivers a good improvements on contact predictions, fold and secondary structure, but interestingly not so much for downstream tasks (specially in Table 8/ Fugire 10 SaAMPLIFY isn’t better than plain AMPLIFY).
SaESM-2 (aligned ESM-2) transfers to downstream tasks better than SaAMPLIFY — again confirming very poor correlation between perplexity and transferability.
ProSST: quantized structure tokens
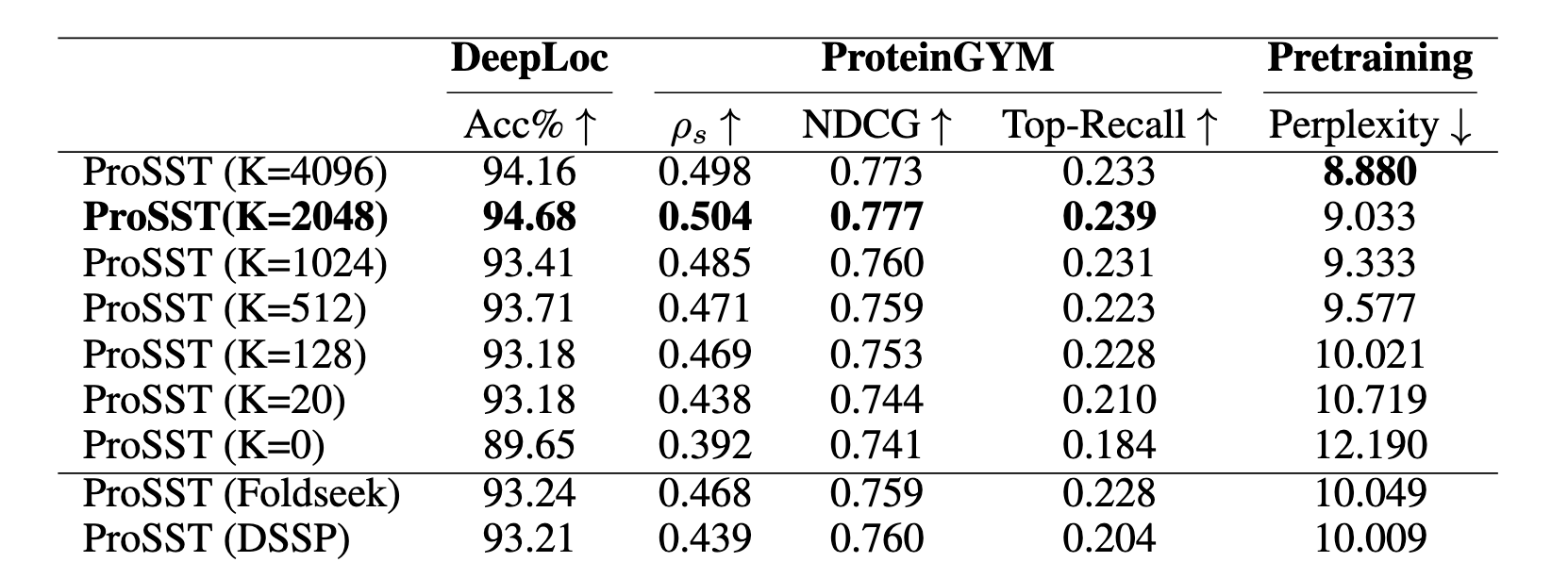
ProSST heads the leaderboard in proteinGYM, let’s see the recipe:
- introduced structure tokens by encoding 40 neighbors
- attention separately encodes sequence, structure tokens and relative position (ablation against the plain attention shows unrealistic improvement, could they have forgotten relpos?)
- pre-trained on AFDB (18.8M structures selected) using ESM-style MLM objective
Result is SOTA generalization to downstream tasks. Peak performance is reached at ~110M parameters, and then goes down.
Model requires knowing the protein structure during prediction, which is somewhat limiting. Huge structural database was used, and perplexity still improves with size, but not downstream performance.
VespaG
VespaG is a tiny projection on top of ESM-2 embeddings, and achieves SOTA performance among sequence-only models. Trick is to “align” token embedding produced by ESM-2 (or other PLM) to MSA-based statistics computed by GEMME.
From their analysis, again, highest performance is reached on 650M ESM2, and then goes down — mirroring results of plain ESM family with some additional boost in quality.
Scaling and Data Saturation in Protein Language Models
Paper starts with a nice reference: in LLM world relation of scaling law and downstream performance is not direct (likely even less so with RL finetuning strategies)
And show how this observation translates to the world of proteins by training a number of AMPLIFY models:
- let’s chunk every sequence. Training on more chunks from same sequences consistently improves performance, while adding newer sequences can hurt it
- When stratifying by MSA depth, proteins with larger MSAs (as measured by Neff/L) tended to show improved prediction performance with later model training years, unlike those with smaller MSAs
- “when partitioning by functional assay type, proteins evaluated using Organismal Fitness as the readout exhibited the most consistent improvement over time, whereas other categories showed more variable or flat trajectories” — this is reasonable, after all nature crafts sequences only by fitness
Finally, an experiment with one specific family shows that supervised dataset can replace a decade of collecting protein data in the wild, so … just collecting sequences-in-the-wild is still useful but inefficient.
Training Compute-Optimal Protein Language Models
Metagenomic sequences are diverse and abundant, likely a good complement to UniProt — so authors add ColabFoldDB in training.
Paper builds a good contrast between MLMs and causal LMs (CLMs). MLMs are efficient and easy to overfit, opposite to CLMs.
They claim that optimal training recipe is starting from CLMs, then switchin loss to MLM; Surprisingly, training on two losses at the same time isn’t better. Authors argue that flops-optimal scaling favors larger models (and they train up to 10B parameters). Results are mixed:
- transfer to downstream tasks isn’t impressive
- contact prediction: minor fine-tuning of ~1B model achieves higher quality than larger model
Insteresting observation: BERT’s 15% masking ratio (used in ESMs) is still a good choice in protein MLMs.
Ankh3: combining sequence denoising and completion
This paper stands out because 1. they show good improvement in contact prediction 2. 6B model is overall better than 2B model.
A model jointly optimized on two objectives: encoder-decoder protein completion and MLM denoising (with 15%, 20% or 50% masking probability, and apparently short spans were masked, not individual tokens). Both points contradict previous paper in this list — could be results of encoder-decoder architecture.
Preprint leaves many questions unanswered:
- model is deep (72 layers), so it could be just ineffecient
- evaluation is limited to datasets without easy ‘leaderboard’ to estimate downstream performance.
- I’m a bit concerned that ESM-2 and Ankh results were “sourced from ankh paper” instead of being reproduced.
ProGen3: Scaling Unlocks Broader Generation and Deeper Functional Understanding of Proteins
- Employ huge curated dataset (PPA-1) that combines genomic and metagenomic sources and excludes fragments.
- Model is trained on left-to-right, right-to-left and span infilling objectives (finally!). Then aligned on downstream tasks using IRPO — modification of DPO.
Results: non-aligned perfomance frequently peaks at ~3B, aligned performance usually still improves. Larger models can generate proteins from more clusters, with tiny implevements in expression.
Exact numbers on proteinGYM aren’t impressive, but overall dynamics after alignment looks encouraging.
DPLM-1 / DPLM-2 / ESM-3
These models were trained with a sufficient amount of structural information in a form of structure tokens.
DPLM-1 achieves better downstream performance on multiple tasks on 3B model (no larger model was analyzed), but DPLM-2 (with primary focus on structure tokens based on LFQ) reports only 650M model — I treat this as implicit signal of scaling boundary. Interestingly, DLPM-2 shows worse downstream performance, and authors link this to missing PLM pretraining in DPLM-2.
Combination of scaling + PLM pretraining + better structure tokens would be very interesting, but this didn’t happen yet with DPLMs (or happened and result wasn’t good enough for publication).
ESM-3 is somewhat close, but they don’t report any actual translatable properties of the model; performance reported by proteinGYM isn’t impressive and ESM-C 300M has similar performance to ESM-C 600M.
MSA as a context for PLMs
MSA-based models (like MsaPairformer) show better transferability compared to PLMs (and they are smaller than PLMs).
PoET model started a direction in PLMs where homologous sequences are passed as a context while architecture is still a classical transformer.
This direction inherits weak sides of both PLMs and MSA-based models: 1. one still has to retrieve MSAs 2. alignment should be done by model implicitly 3. more weights compared to MSA-based models and 4. long+deep MSAs are expensive because of quadratic attention.
One paper from this family (Profluent E1, also trained on PPA-1) claims good perfomance on gym and contact prediction (better than MsaPairformer and other PLMs) and shows positive scaling … up to 600M. From plots I’d expect further improvement on contact prediction, but not on downstream tasks. Given cost of training, it isn’t surprising that largest model is only 600M.
Final thoughts / directions
Multiple years of research in PLMs did not bring a recognized recipe to utilize vast sequencing data. Recent literature contains some interesting hints, but not strong hypotheses how to do this. PLMs more and more incorporate structural or MSA features, which pushes performance; model sizes still mostly don’t matter.
PLMs started from assumption that better perplexity means overall better understanding of protein sequences, as it worked in NLP. This assumption is wrong, and likely in NLP it isn’t true either: longer training on natural language worked because pretty much any reasonable problem was already discussed with examples in the training data. Later progress in NLP was guaranteed by numerous problem-oriented curated datasets, scaling only helped in storing knowledge/patterns in the model.
If, in addition to protein sequences, training data contained various tokens related to expression, function, interaction, biophysical properties, etc., then all those metrics would go up. Protein sequences alone don’t provide enough training signal. Can correlation with other genes from the same organism provide a more useful context? Can functional descrption form a better prompt? Some teams work on this, so we’ll see soon.
Is there a double descent in biology? Given the size of ESM-3 I’ll put this hypothesis off the table.
Are we memorizing phylogenetic noise? Almost surely yes. Larger models can generate proteins from more families (as shown by E1), while the best property prediction is still provided by analysis of MSAs (within the same family).
Maybe nature does not care much about our downstream tasks. Maybe much memory isn’t necessary to memorize everything useful in biology (we’re far from optimal performance, so probably not).
Simple but likely more fruitful direction at this point would be to curate a large dataset with diverse downstream properties.
Confounding factors? We don’t accept assay results at face value, but we generally assume that protein sequences are free of confounding effects (except for phylogenetic noise). In “Clever Hans in Chemistry” authors show that models can guess the author of molecule; knowing author, they can guess the activity without looking at the molecule itself. Could similar cues appear in non-frequent sequences? Like the sequencing technology, or assembly method? This is yet another hypothesis why we don’t see generalization.
 Gradient boosting
Gradient boosting  Hamiltonian MC
Hamiltonian MC  Gradient boosting
Gradient boosting  Reconstructing pictures
Reconstructing pictures  Neural Networks
Neural Networks